Enantioselective catalysis
 Enantioselective catalysts produce selectively either right-
or left-handed molecules. Right- and left-handed molecules
have identical physical properties, but interact differently
with living organisms. Often a left handed drug is
beneficial, while the right handed drug, otherwise
identical, may be even toxic and vice versa. Thus
enantioseletive catalysis is of enormous importance for
pharmaceutical, cosmetic and agricultural industry. The
enantioselective catalysts are relatively complex (about 100
atoms) molecules that contain one or few transition metals
as their catalytic center. These catalysts have themselves a
given handedness and transfer this information to the
chemical reaction.
Enantioselective catalysts produce selectively either right-
or left-handed molecules. Right- and left-handed molecules
have identical physical properties, but interact differently
with living organisms. Often a left handed drug is
beneficial, while the right handed drug, otherwise
identical, may be even toxic and vice versa. Thus
enantioseletive catalysis is of enormous importance for
pharmaceutical, cosmetic and agricultural industry. The
enantioselective catalysts are relatively complex (about 100
atoms) molecules that contain one or few transition metals
as their catalytic center. These catalysts have themselves a
given handedness and transfer this information to the
chemical reaction.
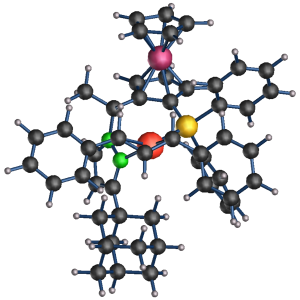
In this project we explored the steps necessary for the rational molecular design of such catalysts. While a number of variants of a catalyst have been explored, their reactivity and selectivity could not be rationalized. The question was if the shape of the catalyst was responsible for the selectivity or if it was due to a tuning of the chemical binding to the catalytically active transition metal center.
 From a
theoretical point of view this study was a major challenge
as it was probably the first ab-initio analysis of a
enantio-selective catalytic mechanism. The energy difference
between the two alternative reaction pathways are of order
few kJ/mol, which is less than the error bar on our
calculations. Was it nevertheless possible to provide
reliable answers?
From a
theoretical point of view this study was a major challenge
as it was probably the first ab-initio analysis of a
enantio-selective catalytic mechanism. The energy difference
between the two alternative reaction pathways are of order
few kJ/mol, which is less than the error bar on our
calculations. Was it nevertheless possible to provide
reliable answers?
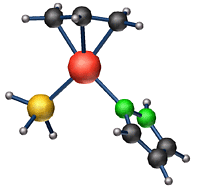
![]() In our approach we exploited the
possibilities of our "theoretical laboratory". By removing
all the ligands we could exclude all shape effects and study
the electronic processes per se. Experimentally, this would
be difficult, because the catalyst would be explosive. We
learned that by rotating the molecule on the catalyst we
could truely switch from one alternative for the reaction
pathway to the other. This finding was robust and did not
suffer from our error bar.
In our approach we exploited the
possibilities of our "theoretical laboratory". By removing
all the ligands we could exclude all shape effects and study
the electronic processes per se. Experimentally, this would
be difficult, because the catalyst would be explosive. We
learned that by rotating the molecule on the catalyst we
could truely switch from one alternative for the reaction
pathway to the other. This finding was robust and did not
suffer from our error bar.
The rotation itself had to be due to a shape effect the ligands. We investigate the complete pair of catalyts and reactand. The system behaved very different from the "small systems" studied so far. As it is known from biological macromolecule the structure developed in steps hopping from one metastable state to the next. Using a ab-initio molecular dynamics and simulated annealing we could bring the system into its stable configuration.
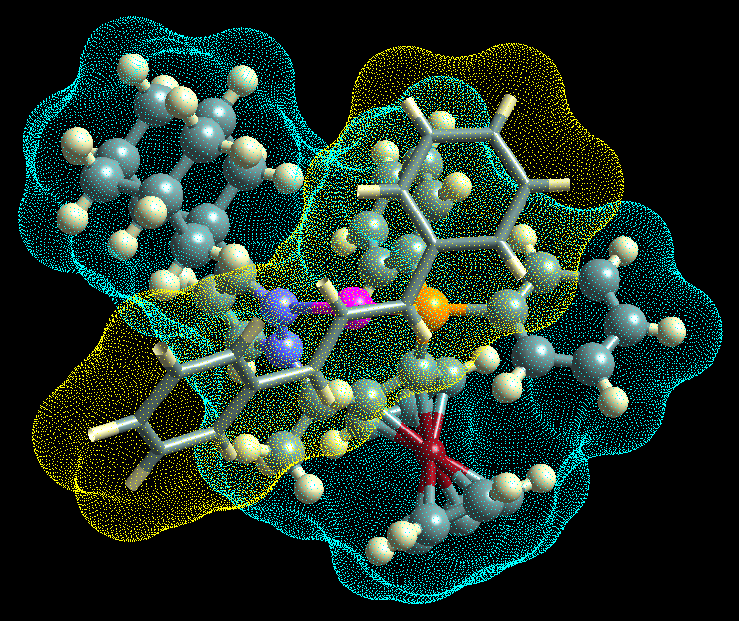 In the stable configuration the reacting molecule cushioned
itself into a valley in the surface of the molecule. This
valey was oriented at an angle to the axis of the active
site, thus explaining the origin for the rotation from the
the main axis of the active site.
In the stable configuration the reacting molecule cushioned
itself into a valley in the surface of the molecule. This
valey was oriented at an angle to the axis of the active
site, thus explaining the origin for the rotation from the
the main axis of the active site.
Thus we could explain the selectivity by a interplay between electronic effects and shape effects, where the latter stear the former. Considering various conformers the different experiments could be rationalized within one common picture. Furthermore the study encouraged us not to give up immediately, if the accuracy does not seem sufficient. Sometimes, a critical analysis allows to overcome these limitations.
For further details see:
"First-Principles Investigation of Enantioselective Catalysis: Asymmetric Allylic Amination with Pd Complexes Bearing P, N Ligands",
P.E. Blöchl and A. Togni, Organometallics 15, 4125 (1996).