How nature breaks the strongest bond -- the workings of the enzyme nitrogenase
Nitrogen is one of the main building materials for biological organisms. Unlike carbon, oxygen and hydrogen, nitrogen it is extremely hard to collect. While nitrogen makes up almost 80% of our atmosphere, there are hardly any minerals containing nitrogen. Nitrogen molecules in the atmosphere are extemely stable and unreactive. During evolution, only certain bacteria have developed the ability to harvest nitrogen from the atmosphere and convert it into ammonia that can be metabolized further. These bacteria posess an enzyme that manages to break one of the strongest bonds in nature. How this is accomplished, is the central topic of this project.
 The enzyme
that performs this conversion is
called
nitrogenase. It is one of the most complex enzymes in nature. The
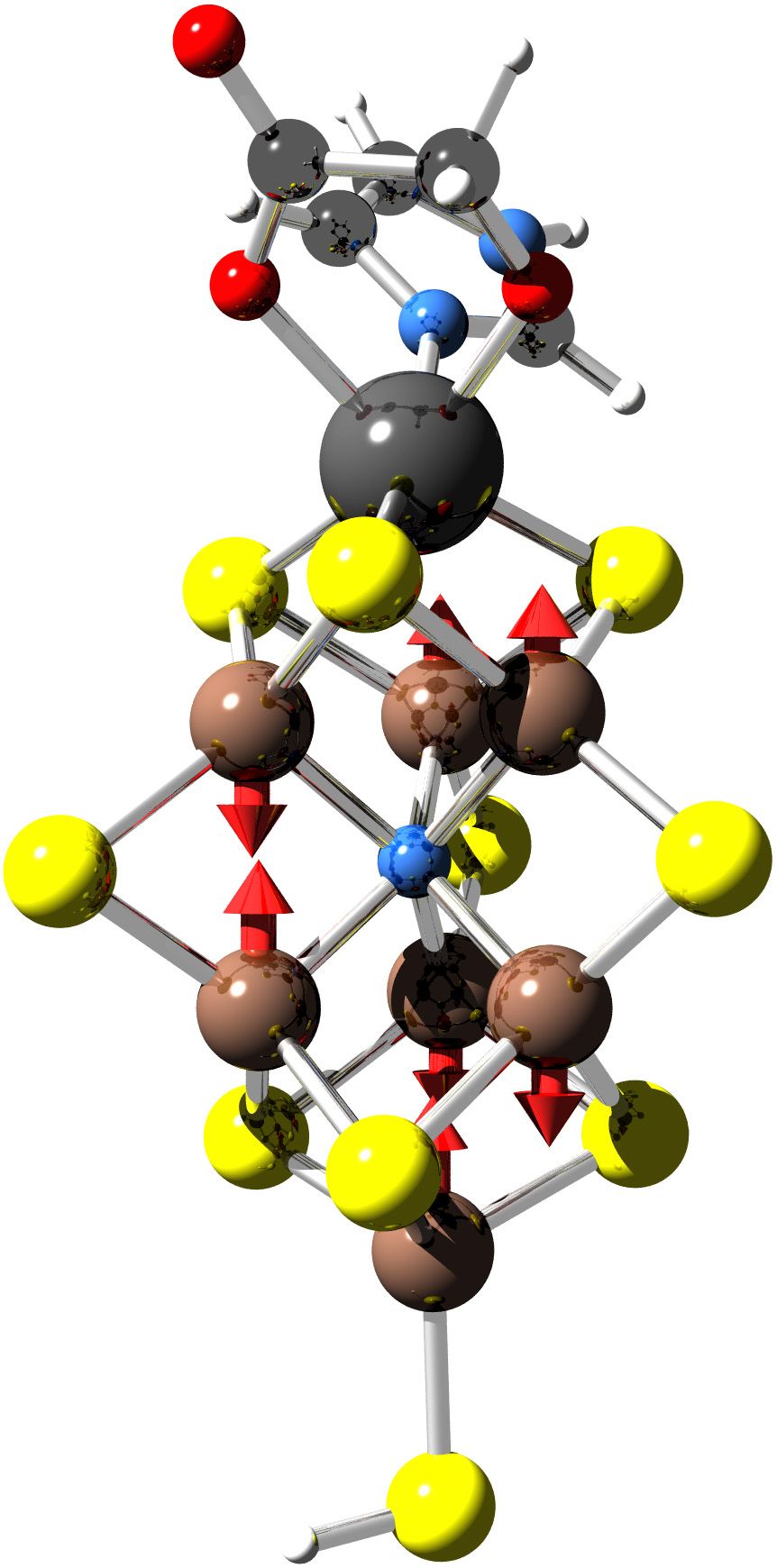
active site is a iron-molybdenum-sulfur cluster, which is called
FeMo-cofactor. Even though the active site of the enzyme has been
unraveled already in the early 1990s, the structure did not provide
sufficient clues on its function. This was the motivation for us to
investigate the function using first-principles simulations.
The enzyme
that performs this conversion is
called
nitrogenase. It is one of the most complex enzymes in nature. The
active site is a iron-molybdenum-sulfur cluster, which is called
FeMo-cofactor. Even though the active site of the enzyme has been
unraveled already in the early 1990s, the structure did not provide
sufficient clues on its function. This was the motivation for us to
investigate the function using first-principles simulations.
Early in our project, which began in 1995, it became evident that this molecule provides an enormous challenge to electronic structure calculations: Its magnetic properties lead to a huge number of configurations. Interestingly, we observed that the cluster liked to suck up a nitrogen atom if a nitrogen molecules is hydrogenated on its surface. Later, this was indeed found experimentally. These finding were exciting, but the situation was still unsatisfactory because it was not clear how reliable our results would be. Thus we extended our simulation tools and implemented so-called non-collinear description of magnetism, where the magnetisation can point not only along one common axis, but in any direction. This required a major overhaul of our code.
Before we could start investigating the workings of the enzyme, we still had to determine the nature of the resting state of the enzyme. Only this state is experimentally accessable. Experimentally one cannot determine the charge of the cluster, but one has seen a characteristic spin signal and one observed structural changes upon charging. By exploring the behavior of different charge and spin states, we could identify the resting state.
From this starting point we carefully worked our way step-by-step through the reaction process. In its resting state the cofactor does not bind nitrogen. Two additional electrons and two protons are needed at the cluster before the nitrogen molecule docks to the cofactor. Electrons and protons arrive alternatingly to avoid a large charge buildup at the cluster. At this point we were ready to explore the reaction cycle.
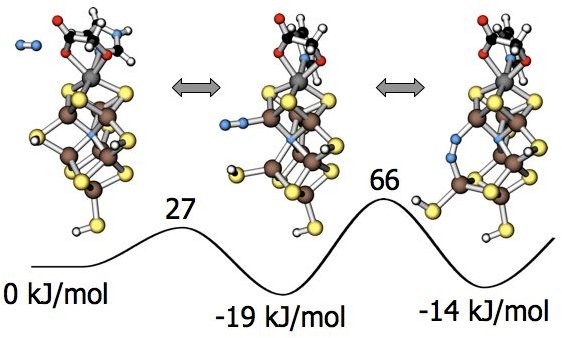
Initially we were poking around to understand how the cluster behaves. Then we faced the ultimate question, namely if one would be able to track a reaction mechanism of such a complex system. Our solution was to turn the question around. The important is not how the reaction goes, but how it does not go. Whatever is left over contributes to the reaction mechanism. Still this is a daunting challenge. Seemingly trivial, this shift in perspective radically changed the approach of the problem. Barriers that prevent reactions are usually high and less sensitive to numerical inaccuracies. The new approach provides an overview of the configuration space and thus a much deeper insight into what actually matters for the reaction. This is invaluable for the design of analogous catalytic systems.
The catalytic cycle of nitrogen fixation
The picture that emerged from our study is collected in the figure shows all the intermediate steps of the catalytic cycle: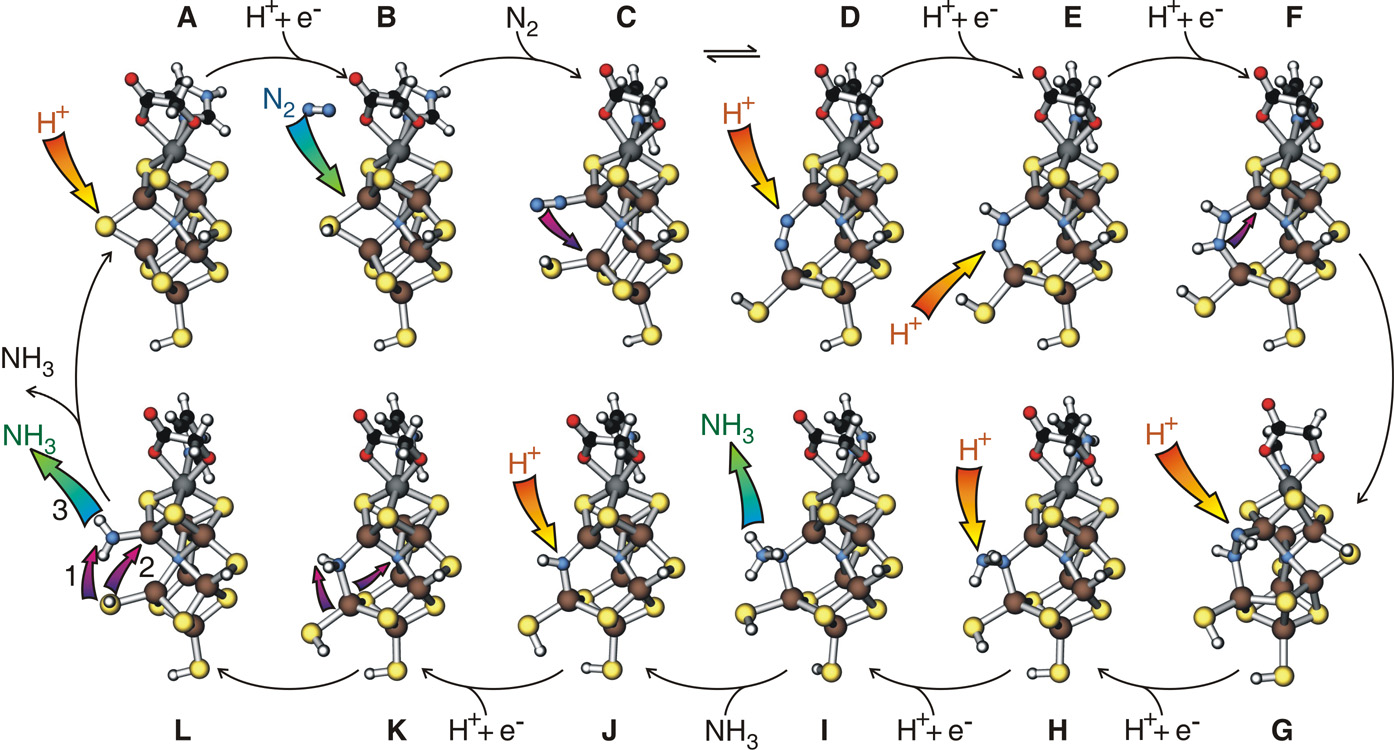 The particular observations of interest are the following:
The particular observations of interest are the following:
-
 The mechanism is a 12-step catalytic cycle. We find, that the cofactor
does not only provide a template for the reaction but that the cluster
opens up and thus takes actively part in the reaction. As dinitrogen
binds, a sulfur bridge between two iron atoms opens up, which allows
the molecule to bind end-on with both of its atoms to Fe ions.
The mechanism is a 12-step catalytic cycle. We find, that the cofactor
does not only provide a template for the reaction but that the cluster
opens up and thus takes actively part in the reaction. As dinitrogen
binds, a sulfur bridge between two iron atoms opens up, which allows
the molecule to bind end-on with both of its atoms to Fe ions.  It has been known that the addition of the first two hydrogen atoms is the
most difficult step in the reaction. The cofactor facilitates this
reaction by establishing alread to weak bonds of the nitrogen molecule
to its iron atoms. With two bonds already present the formation of
additional bonds proceeds much easier.
It has been known that the addition of the first two hydrogen atoms is the
most difficult step in the reaction. The cofactor facilitates this
reaction by establishing alread to weak bonds of the nitrogen molecule
to its iron atoms. With two bonds already present the formation of
additional bonds proceeds much easier.Interesting is also the last step. A catalyst is said to bind the transition state, thus stabilizing the most unfavorable configuration along the reaction. However, if a catalyst does this too well, it runs the danger of also holding on to the reaction products. Being cluttered with reaction products the catalyst cannot accept a reactand for the next cycle: Additional nitrogen molecules cannot bind. Nitrogenase gets rid of the second ammonia, by closing a sulfur bridge between to iron atoms, kicking the second ammonium away from its binding site.
A long-standing puzzle has been the hydrogen production. During the reaction also a hydrogen molecule is formed. As hydrogen production is an extremely wasteful process it was believed that it was due to an essential step in the reaction cycle. Our calculations show that hydrogen production is merely an inefficiency of the enzyme. In order to bind nitrogen, the cluster has to accumulate so many electrons and protons. If a proton arrives at the binding site for nitrogen first, it will simply join with a nearby proton, grab two electrons from the cluster and disappear as hydrogen molecule. This picture consistent with experimental findings about the competition of nitrogen fixationand hydrogen production.
For further details see
- Ammonia Production at the FeMo-Cofactor of Nitrogenase: Results
from Density-Functional Theory,
J. Kästner and P.E. Blöchl, J. Am. Chem. Soc. 129, 2998 (2007) - "Activation and Protonation of Dinitrogen at the FeMo-Cofactor
of Nitrogenase",
Johannes Kästner, Sascha Hemmen and Peter E. Blöchl, J. Chem. Phys. 123, 74306 (2005). - A Model for Acetylene Reduction by Nitrogenase Derived from
Density Functional Theory,
J. Kästner and P.E. Blöchl, Inorg. Chem., 44, 4568 (2005). - "Towards an understanding of the workings of nitrogenase from
DFT calculations",
, J. Kästner und P.E. Blöchl, ChemPhysChem 6, 1724 (2005). -
Nitrogen Binding to the FeMo cofactor of Nitrogenase,
Johannes Schimpl, Helena M. Petrilli, and P.E. Blöchl, J. Am. Chem. Soc. 125, 15772-15778 (2003).